Deep learning the cis-regulatory code for gene expression in selected model plants - Nature Communications
🌈 Abstract
The article explores the relationship between non-coding regulatory element sequences and gene expression in plant species, using deep learning models to predict gene expression profiles from gene flanking regions. The models achieved over 80% accuracy in predicting high and low gene expression, and enabled the identification of important regulatory sequence features, particularly in the UTR regions. The models demonstrated cross-species performance, highlighting both conserved and species-specific regulatory sequence features. The approach was applied to reveal links between genetic variation and gene expression changes across tomato genotypes, and to predict genotype-specific expression of key functional gene groups.
🙋 Q&A
[01] Elucidating the relationship between non-coding regulatory element sequences and gene expression
1. Questions related to the content of the section?
- What is the key objective of the study?
- What are the main methods used to explore the relationship between regulatory sequences and gene expression?
- What are the key findings regarding the predictive power of the deep learning models for gene expression?
The key objective of the study is to systematically identify gene regulatory sequences and annotate their function in terms of their effect on gene expression, across multiple plant species. The main methods used are:
- Training convolutional neural network (CNN) models to predict gene expression states (low, medium, high) from the DNA sequences of gene flanking regions.
- Using the trained models to identify predictive sequence features (expression-predictive motifs or EPMs) and their conservation across species.
- Demonstrating the application of the models to annotate genetic variation and predict metabolic pathway activity in different tomato genotypes.
The key findings are:
- The CNN models achieved over 80% accuracy in predicting high and low gene expression from the gene flanking sequences.
- The models were able to identify important regulatory sequence features, particularly in the UTR regions, that are predictive of gene expression levels.
- The models showed good cross-species performance, indicating conservation of key regulatory sequence features across the plant species studied.
- The approach was successful in revealing links between genetic variation and gene expression changes, and in predicting genotype-specific expression of functional gene groups.
[02] Model architecture and training strategy
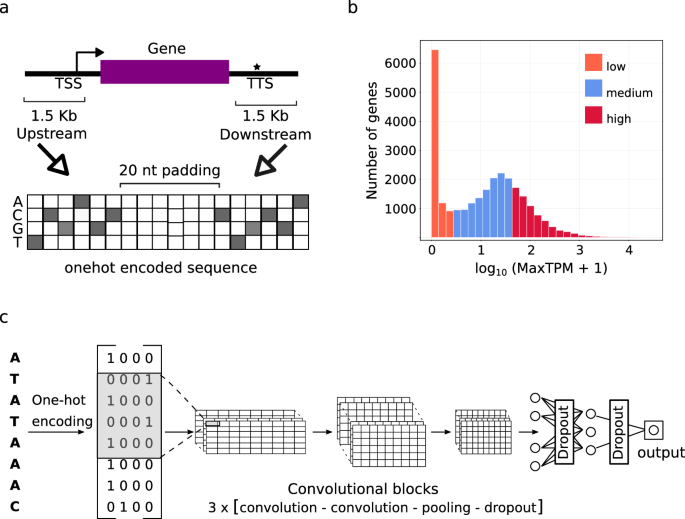
1. What was the CNN model architecture used in the study? The CNN model architecture consisted of three convolutional blocks, each with two convolutional layers, followed by a fully connected block. 1D convolutional and max pooling layers were used, as opposed to 2D layers used in previous studies.
2. How were the training and validation sets constructed? The training and validation sets were constructed using a chromosomal level cross-validation approach, where genes on one chromosome were used as the validation set and the rest for training. To prevent overfitting due to evolutionary relatedness, genes in the validation set that had homologs in the training set were excluded.
3. What were the key findings regarding model performance?
- The single-species reference (SSR) models achieved average accuracies of 79-86% for predicting high vs low expressed genes.
- The multi-species reference (MSR) models, trained on a combination of species, showed similar performance to the SSR models, indicating conservation of predictive sequence features.
- The cross-species prediction performance was lower than the SSR and MSR models, suggesting the existence of both conserved and species-specific regulatory sequence features.
[03] Identification and characterisation of predictive sequence features
1. What were the key findings regarding the predictive sequence features (EPMs) identified by the models?
- The most salient regions were the proximal sequences around the TSS and TTS, containing the 5' and 3' UTRs.
- The EPMs exhibited both conserved and species-specific patterns in terms of sequence and positional preferences.
- Many of the EPMs showed significant similarity to known transcription factor binding sites, while others corresponded to structural gene elements like polyadenylation signals.
- Some EPM clusters, like 2CWY+ and 2GCB+, showed consistent positional preferences and predictive power across species.
2. How were the EPMs further characterized and their predictive power evaluated?
- The EPMs were divided into two metaclusters based on their positive or negative importance scores, corresponding to prediction of high or low expression.
- The occurrence of EPMs from the high expression metacluster was found to be a strong predictor (>80% TPR) of high gene expression in Arabidopsis.
- In contrast, the EPMs from the low expression metacluster were not as enriched in the low expressed genes, suggesting their predictive power may depend more on contextual factors.
[04] Application to tomato genotypes and metabolic pathways
1. How was the approach used to analyze differences in gene expression between tomato genotypes?
- The MSR model trained on multiple species was used to predict gene expression differences across 14 diverse tomato genotypes.
- Genes with predicted differential expression between genotypes were found to have mutations in the EPM regions, coinciding with observed expression changes.
- Two example genes were analyzed in detail, showing how mutations in EPM regions can impact the model's expression predictions.
2. How was the approach used to highlight differences in metabolic pathways between tomato species?
- The MSR model was used to predict expression of genes in the polyamine biosynthesis pathway between cultivated tomato (S. lycopersicum) and its wild relative (S. pennellii).
- The model was able to correctly identify the most likely highly expressed isoforms for each pathway step, and highlight differences in expression of orthologous genes between the two species.
- This demonstrated the model's ability to efficiently prioritize candidate genes for further experimental validation of metabolic differences between related plant species.